In order to ensure that ERCC data is of a high standard we have developed quality control (QC) standards for extracellular RNA-Seq data being generated by the consortium. Since the experimental protocols for these experiments are still being optimized, these standards will evolve over the duration of consortium. These initial standards (version 1.0) are developed to be somewhat inclusive and to identify experiments that are clear outliers. The initial plan is that all initial experiments submitted to the DCC will be displayed in the exRNA Atlas. Datasets that do not meet the quality control standards will be flagged but not removed.
In order to develop QC standards, we uniformly processed 595 small exRNA-Seq datasets through version 3.2.6 of the exceRpt small RNA pipeline. These samples were generated by 7 different ERCC labs using a variety of different RNA isolation methods, library preparations, and biofluids. The following two QC standards were developed following discussions on the MADS, RNA-Seq, and U01 reference profiling working groups as well as discussion at the 3rd ERC Consortium meeting in Washington DC in November 2015:
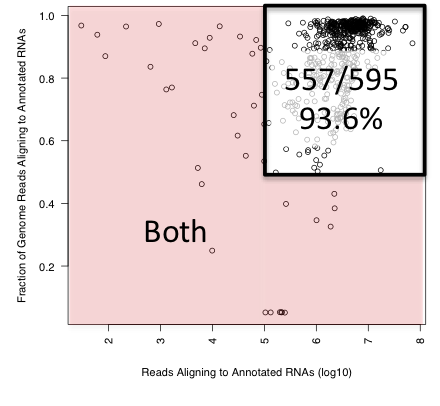
Figure 1: Distribution plot of 595 small RNA-Seq datasets showing reads aligning to any annotated human transcript along the x-axis and the fraction of genomic reads that align to any annotated transcript along the y-axis.
Both of these QC metrics are automatically computed by the exceRpt pipeline for every RNA-Seq dataset processed. The motivation for including any annotated RNA biotype is an acknowledgement that different experimental RNA isolation and library preparation protocols might target different types of RNA, and we would rather start with generic standards for all experiments agnostic to the RNA annotation detected. The first criterion ensures that a sufficient number of reads are generated to accurately identify and quantitate the RNAs present in the extracellular sample of interest. The second criterion was developed to identify samples that have low fractions of genomic reads aligning to annotated RNAs, the likely cause of which is DNA contamination, possibly from cellular sources. Applying both of these criteria to the 595 datasets we find that 557 (93.6%) meet both criteria, with the majority of datasets being comfortably above both thresholds.
These two QC standards are intended to identify and flag small RNA-Seq experiments that are clear outliers. These represent initial standards (version 1.0) that will be refined as more data is generated by the consortium and as experimental protocols are standardized. The exceRpt pipeline computes several additional metrics for each dataset which are not currently used for QC. We will continue to explore the utility of these other metrics for identifying poor quality samples as new data become available.