“Decoding the Data Ecosystem” is a new podcast series dedicated to unraveling the complexities and exploring the depths of omics research. Hosted by Dr. Alissa Dillman, Engagement and Outreach Director in the Office of Data Science Strategy at the NIH.
Listen now to the episodes below, and be sure to subscribe for new ones. Visit the podcast website here.
Episode 1: Unveiling the Strengths of the CFDE: A Resource for All Researchers
Episode 2: Reproducibility and Training on the Elements of Style
Episode 3: Navigating the Common Fund Data Ecosystem’s Data & Resources to Empower Scientific Discoveries
Episode 4: How the Data Resource Center Uses the FAIR Principles to Increase Accessibility and Interoperability

- Learn about the CFDE, an open-access resource enabling broad use of NIH Common Fund Data
- Learn to navigate and use CFDE data, analysis tools, and knowledge
- Learn how CFDE can accelerate common and rare human disease research and basic biological discoveries
Register for the event here, further details are located on this information page. Please circulate it to interested colleagues! We hope to see you there, but we’ll also record and post the workshop for those who can’t make it.
Best regards,
MacKenzie, for the Knowledge Center team
A recording of the September CFDE Workshop is available now on YouTube, link below!
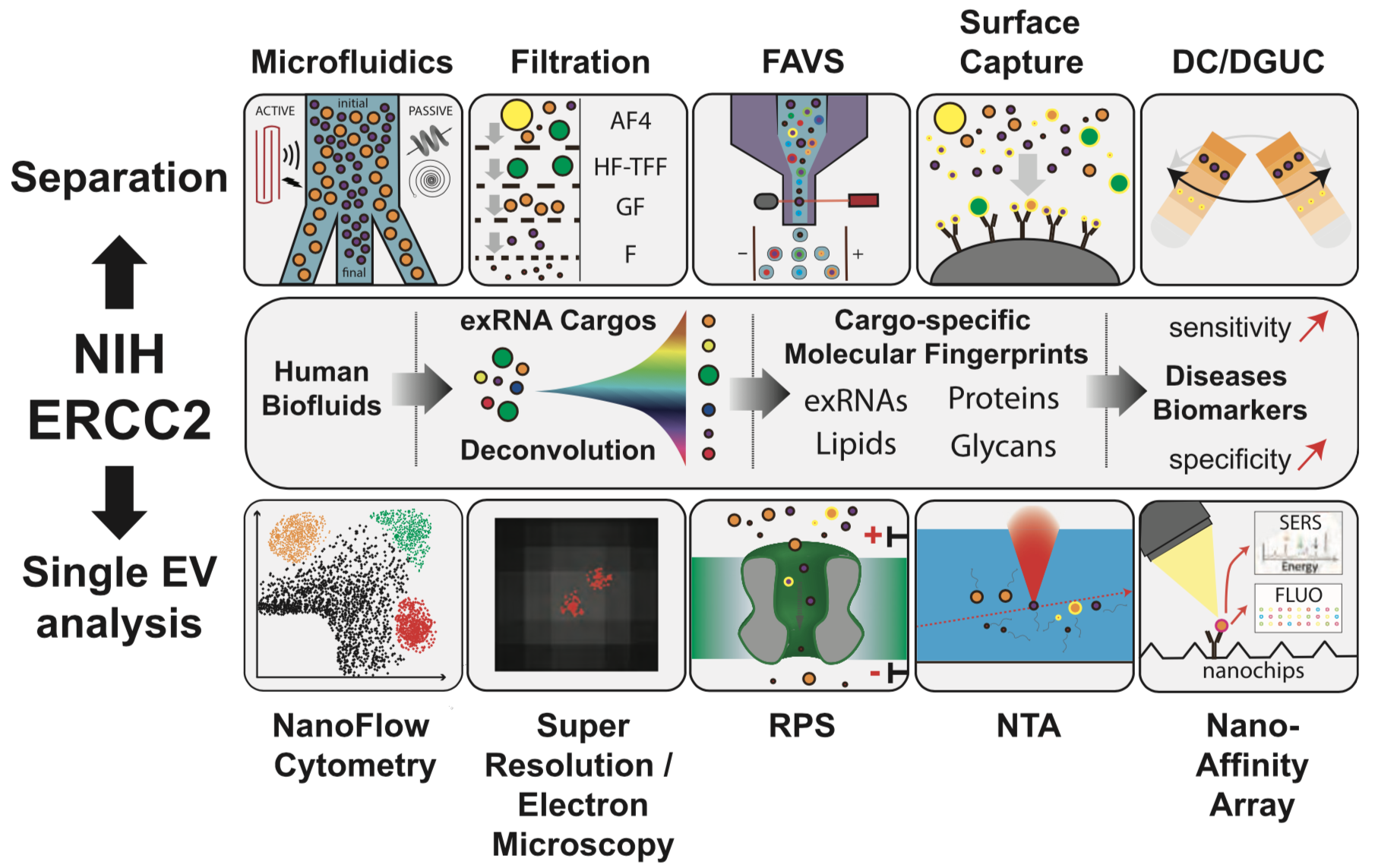
For a description of the workshop please see the original post CFDE Virtual Workshop – exRNA Portal
