Despite being one of the earliest known classes of non-coding RNA molecules, transfer RNAs (tRNAs) are still notoriously difficult to study. The challenge is largely due to this molecule’s secondary structure, chemical modifications to its constituent nucleotides (see figure), and the multiplicity of tRNA genes. As the number of non-coding RNA datasets proliferates, it is becoming increasingly important for tRNA genes to be accurately annotated. In a recent study, Thomas Tuschl from Rockefeller University and colleagues tackled this problem by developing a new protocol for sequencing tRNAs. The new method enabled them to assemble an atlas of human tRNAs for other researchers to use in analyzing their non-coding RNA data.
Hydro-tRNA Sequencing
Transfer RNAs have thermodynamically stable secondary and tertiary structures, and their constituent nucleotides are highly modified by RNA editing. Both of these characteristics are problematic for traditional RNA sequencing methods. The key to the Tuschl lab’s protocol, called hydro-tRNA sequencing (hydro-tRNAseq), is a partial alkaline hydrolysis step that breaks the 60-100 nucleotide-long tRNA into smaller fragments with fewer RNA modifications. These fragments, 19-35 nucleotides in size, have weaker secondary structure and fewer RNA modifications per fragment than the parent tRNA.
Applying the method to short RNA extracted from human embryonic kidney (HEK293) cells resulted in an increase in the fraction of reads mapped to tRNA between 2% and 40%, depending on the depth of sequencing. The short fragment length also improved read accuracy per base compared to standard tRNA sequencing.
To develop a thorough and representative reference set of human tRNAs, the HEK293 dataset was subjected to iterative cycles of mapping to existing reference tRNAs followed by manual curation. In each round, all transcripts with an error distance (number of mismatches, insertions, and deletions) of 1-2 from a given tRNA were kept as candidate reference sequences if they could be attributed to a tRNA isoacceptor (i.e. a different tRNA that binds to the same amino acid). If not, assuming that other mismatches were caused by misidentifying a modified base, transcripts with more than 10% mismatches compared to reference were expanded into a set of all possible combinations of RNA modifications and included in the reference pool (see figure). This mapping and selection process was repeated until there were no longer any modified positions left with a mismatch frequency over 10% compared to reference.
Candidate pre-tRNA genes were obtained by mapping the final tRNA reference sequences back to the genome. Altogether, this analysis was able to account for 93% of the 114 million reads in the deepest library of HEK293 cells’ tRNAs.
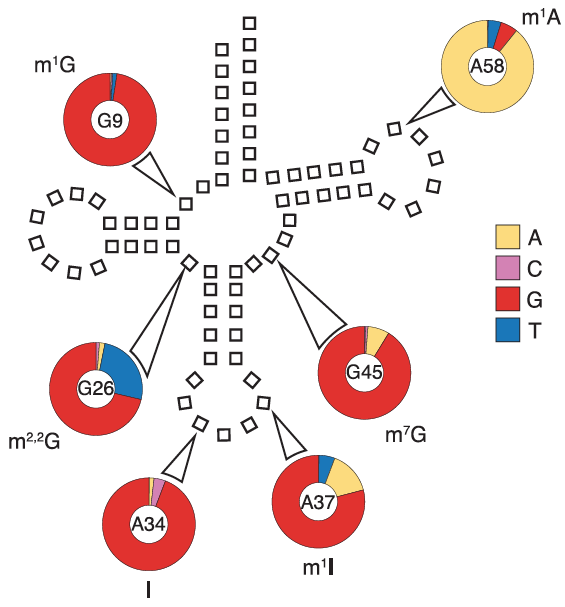
tRNA Modification Sites
The team identified sites of modification from the high frequency of mismatches during mapping caused by read errors there during reverse transcription. Here the reference nucleotide is at ring center, known modification outside the ring, and frequency of each nucleotide read at that site inside the ring.
Source: Cell Reports
The Added Power of SSB PAR-CLIP
Though hydro-tRNAseq greatly improved the reference dataset of human tRNAs, there was still a risk that it alone would miss pre-tRNAs expressed at low levels or processed quickly into mature tRNA. Previous efforts to assay that ephemeral population employed ChIP-seq of POLR3, the polymerase that transcribes all tRNA genes, but doing so assumed that polymerase binding always led to expression and complete processing. The Tuschl lab focused instead on SSB, a protein that binds to the 3′ end of pre-tRNAs, immunoprecipitating tRNAs crosslinked to SSB using a method called PAR-CLIP. As predicted, almost half of the reads from their SSB PAR-CLIP experiments mapped to pre-tRNAs. Combining SSB PAR-CLIP with hydro-tRNAseq allowed the team to better identify mature and pre-tRNAs with improved, accurate, nucleotide-level resolution.
This study supplies the community with several new and useful resources. Hydro-tRNAseq provides a new method to overcome many of the struggles of tRNA sequencing analyses. Combining this method with SSB PAR-CLIP enabled the construction of a comprehensive atlas of pre-tRNAs and mature tRNAs in humans. This methodology can now be applied to study the tRNA complement in other species to further dissect tRNA biology.
Reference
Tasos Gogakos T, Brown M, Garzia A, Meyer C, Hafner M, & Tuschl T. Characterizing Expression and Processing of Precursor and Mature Human tRNAs by Hydro-tRNAseq and PAR-CLIP. Cell Reports (2017) 20: 1463-1475. doi: 10.1016/j.celrep.2017.07.029