This blog originated as a press release from The Ohio State University. Thanks to them for allowing us to repost it here.
Dr. Raphael Pollock has earned the reputation as one of the world’s best surgical oncologists for patients facing one of the toughest cancers to treat, sarcoma. Frequently these tumors start out in the very deepest recesses of the retroperitoneum, the part of the abdomen where the kidneys, pancreas and inferior vena cava are located.
 |
Dr. Raphael Pollock
|
Director of The Ohio State University Comprehensive Cancer Center, Pollock’s 30 years of experience in the operating room naturally led him to ponder the steps before surgery, specifically if there were better ways to diagnose or detect sarcomas. In early 2019, he tapped into Ohio State’s scientific breadth and depth to investigate a new diagnostic method based upon research he conducted while at MD Anderson Cancer Center in Houston. He reached out to Shaurya Prakash, associate professor of mechanical and aerospace engineering and an expert in microfluidics.
Currently, there are two predominant options to acquire a diagnostic biopsy of a tumor deep in the abdomen: invasive surgery under general anesthesia; or a method utilizing computed tomography (CT) scans to guide a long needle through the skin to acquire tissue from the mass. Both are expensive and take time to schedule.
“I’ve been interested for a while in the role of exosomes in the spread of cancers,” Pollock said. Exosomes are extracellular vesicles containing constituents—protein, DNA, and RNA—of the cells that secrete them. They can affect function and behavior of other cells with which they interact. Until recently, they were regarded as merely cell waste products without much clinical research relevance.
 |
Dr. Shaurya Prakash
|
“We learned that there are a number of things inside the exosomes that interact potentially with cells in the tumor microenvironment,” he added. “Then they circulate in the bloodstream and land in other parts of the body.”
So Pollock asked Prakash if there could be an efficient way of extracting these exosomes from a peripheral blood sample to obtain the contents that might be used to diagnose a tumor deep within the body. The microfluidics expert was intrigued.
“I learned that often by the time sarcomas are diagnosed, the disease state is very advanced,” Prakash said. “The value of isolating these circulating biomarkers is earlier detection. Prognosis is better with earlier detection and diagnosis.”
In the past, Pollock had employed ultracentrifugation to isolate exosomes from blood, but it was arduous and expensive. He and Prakash reviewed the literature and realized there might be several different engineering concepts that could be leveraged to improve the process.
Size-based filtration was first, since exosome size is quite specific. Prakash’s previous water treatment research was useful in developing a microfluidic filtration system. Their second area of focus was targeting a surface marker or protein with monoclonal antibodies to attach, secure and extract the exosomes.
 |
Microfluidic device prototype
|
The duo’s prototype microfluidic device integrates size-based separation followed by immunoaffinity-based capture of extracellular vesicles in one process. They also are exploring the use of electrical charge to enhance the exosome filtering.
Prakash and Pollock have submitted two manuscripts—one of which was published recently in the Journal of Microelectromechanical Systems—demonstrating their device is more effective than ultracentrifugation in terms of time, yield, and purity.
The collaboration is just the latest example of an emerging partnership between the College of Engineering and The Ohio State University Comprehensive Cancer Center – James Cancer Hospital and Solove Research Institute.
“These circulating biomarkers are a very small fraction of the overall constituency of blood,” explained Prakash. “The real engineering challenge is extracting that proverbial needle from a haystack. And how do you sort that out and get the right needle.”
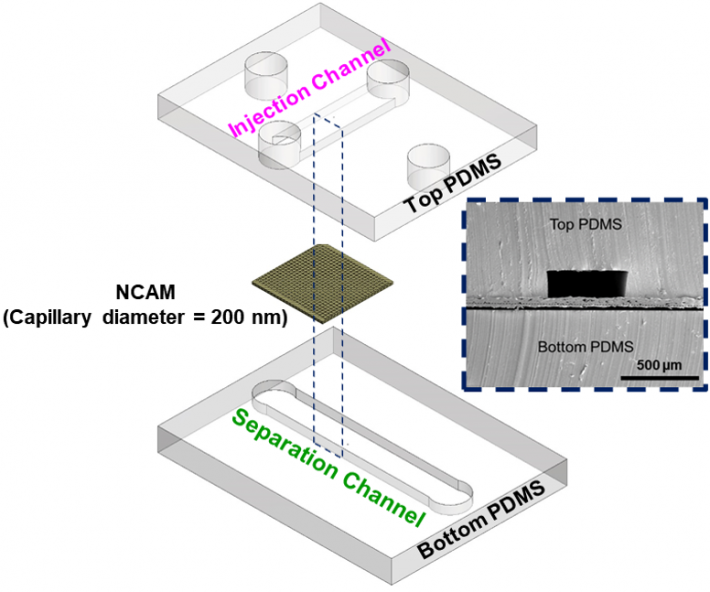 |
Exploded view of microfluidic channels separated by a nanocapillary array membrane. The dotted line represents the cross-section that was utilized for SEM characterization of the device, as seen inset. Only a portion of captured exosomes may have been connected to the tumor, so capturing as many as possible within a blood sample is critical.
|
Beyond the manuscripts, the research team is gearing up to submit a proposal. Coincidentally, the National Cancer Institute is now seeking proposals that focus on developing new methodologies to extract exosomes to investigate whether the cargo inside may be applicable as biomarkers for cancer.
“We’re now in a position to really drill down into the engineering concepts of the device,” Pollock said.
He added that while this type of device could be applied to many types of cancers, it is especially advantageous for sarcoma diagnosis.
“After a long operation to remove a confirmed tumor, it is very difficult in scans to differentiate tumor recurrence from post-surgical scarring,” he explained. “But if you can detect something a tumor releases in the bloodstream, that provides you with a higher index of suspicion of what you may be seeing on a scan.
“Instead of relying on repeat scans over months to determine size increase or decrease, we can potentially identify recurrence at a very early point when the total volume of recurrence is small and more amenable to treatment. We’re very excited about the potential.”
Looking ahead, Prakash and Pollock want to build toward a systematic clinical trial. While there is nothing in the prototype device that cannot be used in a clinical trial, Prakash said some optimization would be required.
“It’s been a total partnership,” Pollock said. “None of this would have happened without the mutual interest and opportunities to communicate about possibilities.”
The research team included mechanical and aerospace engineering PhD student Prashanth Mohana Sundaram, and Lucia Casadei, Gonzalo Lopez, Danielle Braggio and Gita Balakirsky from the Comprehensive Cancer Center.