Saliva is a highly desirable body fluid for biomarker development for clinical applications as it provides a non-invasive, simple and low-cost method for disease detection and screening. Disease detection from a saliva sample has been addressed as one of the so-called Grand Challenges of the 21st century in President Barack Obama’s Strategy for American Innovation. In the last decade, the potential use of salivary RNA has been demonstrated for detecting various local and systemic diseases such as oral cancer, Sjögren syndrome, pancreatic cancer and breast cancer. We believe that the ability to characterize salivary exRNA with next-generation sequencing can further strengthen the advantages of using saliva as a clinical diagnostic biofluid for biomarker discovery.
Compared with other biofluids, saliva can be collected easily and noninvasively. However, low RNA abundance, small sample volumes, highly fragmented RNA and high abundance of bacterial contents create challenges for downstream RNA sequencing assays. Our experience in the field reveals that salivary exRNAs need different processing methods from other biofluids. RNA extracted from cell-free saliva (CFS) from low speed spinning contains significant levels of bacterial RNA. A subsequent cell removal step with high speed centrifugation resulted in all of the intact ribosomal RNAs (rRNA) being detected by Bioanalyzer in the pellet rather than in the cleared supernatant (SN). This observation, combined with the migration of the rRNA peaks (which is more rapid than typically seen for eukaryotic 28S and 18S rRNAs) support the notion that these rRNAs are of bacterial origin (Figure 1, SN & pellet bioanalyzer profile). These findings demonstrate that rRNA contamination, which for most biofluids would be presumed to be of cellular origin, was likely due to the high bacterial load in saliva. This conclusion was further proved by analysis of the resulting NGS data. Only 6-14% of reads mapped to the human genome and 60-70% of reads were from the microbiome, with the majority of sequences representing bacterial rRNAs. These results indicate the need to develop additional technical capabilities to rid samples of microbial rRNA sequences where the final goal is to ascertain the comprehensive salivary exRNA profile.
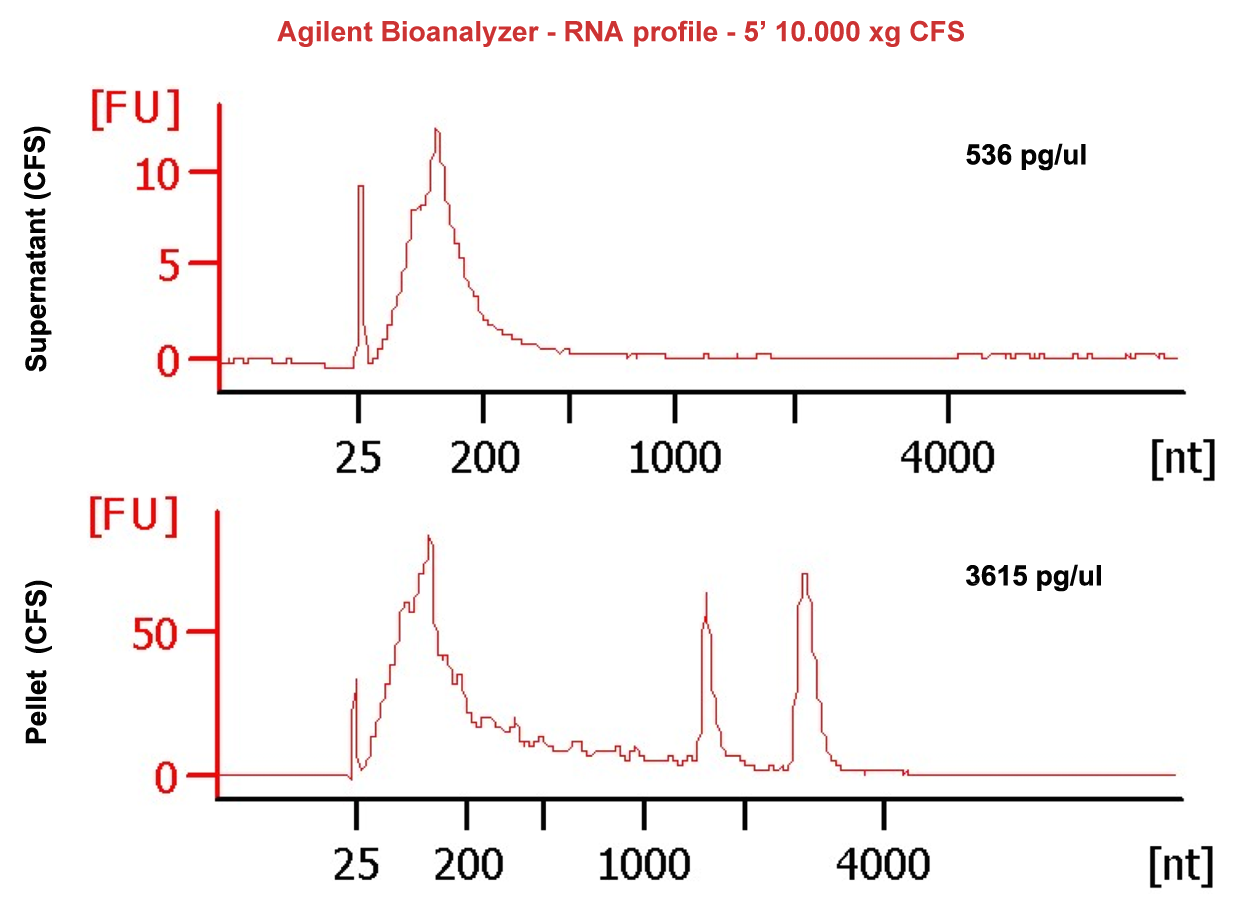
Figure 1: SN & pellet bioanalyzer profile
We have systematically tested RNA isolation efficiency with six commercially available kits with optimized protocols and compared their performance on mimic clinical saliva samples. We compared the RNA yield from saliva samples using six RNA isolation methods: (1) organic extraction method (Trizol LS); (2) spin filter based method (QIAamp Viral (Qiagen), NucleoSpin (Clontech) and miRVana (Life Technologies)); and (3) combined method of organic extraction and spin filter clean up (miReasy micro (Qiagen), Quick-RNA micro (Zymo). The quantity and size distributions of the resulting RNA samples were assessed using the RiboGreen reagent and the Bioanalyzer, respectively (Figure 2, Kits comparison), with the best yields from the NucleoSpin and miRNeasy micro kits.
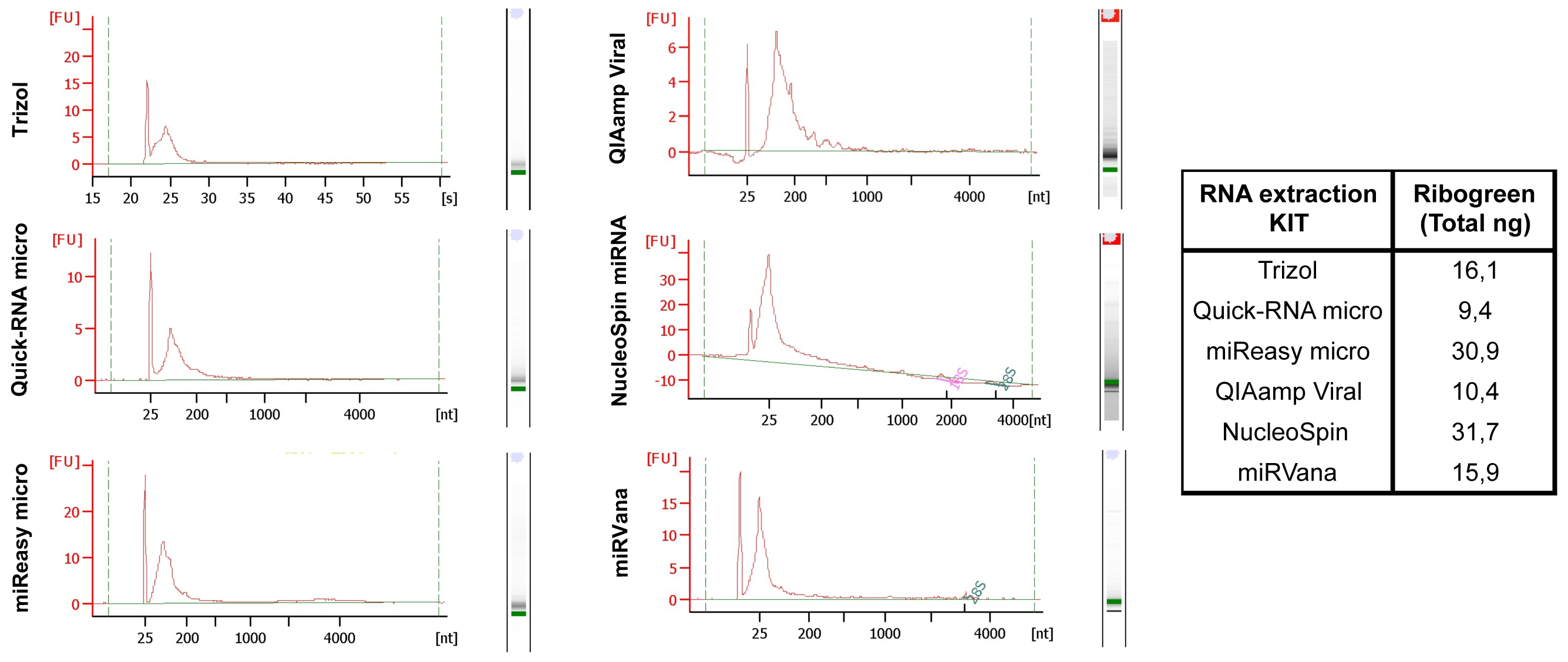
Figure 2: Kits comparison
Quantification of exRNAs is particularly challenging, given that they are typically at low concentrations and have a wide range of lengths, with a prominent population of small RNAs. It is important to keep in mind that different measurement techniques will yield very different total amounts of RNA. Agilent Bioanalyzer profile (Eukaryotic RNA Pico Chip Bioanalyzer) and High sensitive Ribogreen reagent were used as quality controls (QCs) to evaluate the quality and quantity of total RNA yield, respectively. The Ribogreen reagent (available as the Quant-iTTM RiboGreen RNA Assay Kit and Reagent, Life Technologies) can be used to quantify samples at a concentration as low as 50 pg/ul, but is affected by the presence of DNA since it binds to DNA as effectively as it binds to RNA. Although this method can be used to quantify RNA, only the Agilent Bioanalyzer, which has a lower range limit of 50 pg/ul for the RNA Pico and Small RNA Chips, can evaluate the integrity of the RNA molecules. However, this method is not as reproducible in terms of RNA quantification as Ribogreen-based assays and does not distinguish between RNA and DNA. The Bioanalyzer methods are also affected by impurities that can quench the fluorescent signal. Variability in the height of the internal marker peak, an uneven baseline, and an imperfect size standard ladder are indicators that there may be factors present that compromise the accuracy of Bioanalyzer quantification. Therefore, it is important to consider the characteristics of each quantification method, in terms of the limit of detection, dynamic range, and specificity for nucleic acid type, so the most accurate method can be used for the expected yield of RNA, since the measured total RNA yield will vary based on the quantification method used (Figure 3, Ribogreen vs Bioanalyzer quantification).

Figure 3: Ribogreen vs Bioanalyzer quantification
With all these factors in mind, we set up several thresholds for the QCs that the exRNA saliva samples should achieve to go further with the NGS analysis. Thus, all the samples should yield higher than 5 ng of total RNA measured by Ribogreen (RNA yield is about 20-80 ng/mL), and no intact ribosomal RNA peaks should appear in the Bioanalyzer profile.
Besides the RNA isolation and quantification setup, we performed qPCR/ddPCR assays to determine the efficiency of long and small RNA isolation from each kit, showing that RNeasy micro Kit and NucleoSpin are the best kits in yielding small RNAs and long RNAs simultaneously (Figure 4, ddPCR/qPCR data).
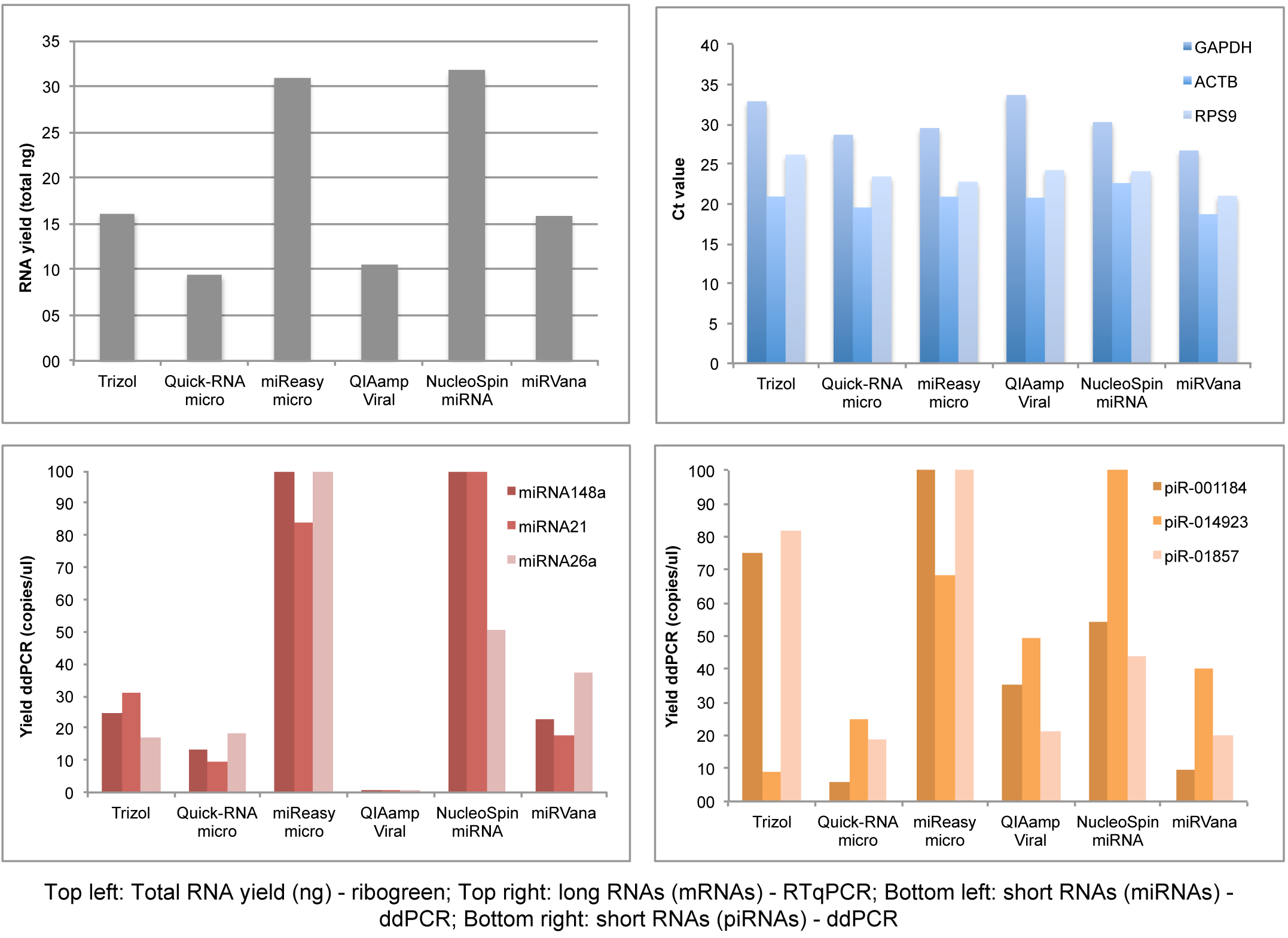
Figure 4: ddPCR/qPCR data
Among the commercially available RNA-Seq library construction methods that we tested, the NEB library preparation kit resulted in the highest number of human genes and small RNAs species. Generally, 23-36% of reads from long-RNA libraries are from bacterial ribosomal RNA. We tried to increase the sensitivity to human transcripts by using a protocol to selectively remove bacterial rRNA (Ribo-Zero(TM) Magnetic Kit for Bacteria, Epicentre). We tried several conditions, combining different proportions of biotinylated beads and rRNA-depletion-probes, to maximize rRNA depletion while avoiding the loss of human species by overloading the reaction with too many beads and/or probes. The mapping results showed many fewer microbial reads (0.8%-4.6%), and at least 48% more genes can be detected in rRNA-depleted samples compared to the control (Figure 5, rRNA-seq results).

Figure 5: rRNA-seq results
This indicates the rRNA removal step could improve the comprehensiveness of human exRNA profile in saliva. Although the rRNA levels were reduced around 95%, we found a high loss in total RNA yield in the depleted samples compared to the controls (checked by Ribogreen assay). We found higher numbers of human genes in the depleted samples, which means that we were able to go deeper in the sequencing process, but we observed a reduction in quantitation for several mRNA, miRNAs and piRNAs in the depleted samples compared to the controls (Figure 6, Total yield, qPCR/ddPCR depleted vs control samples).
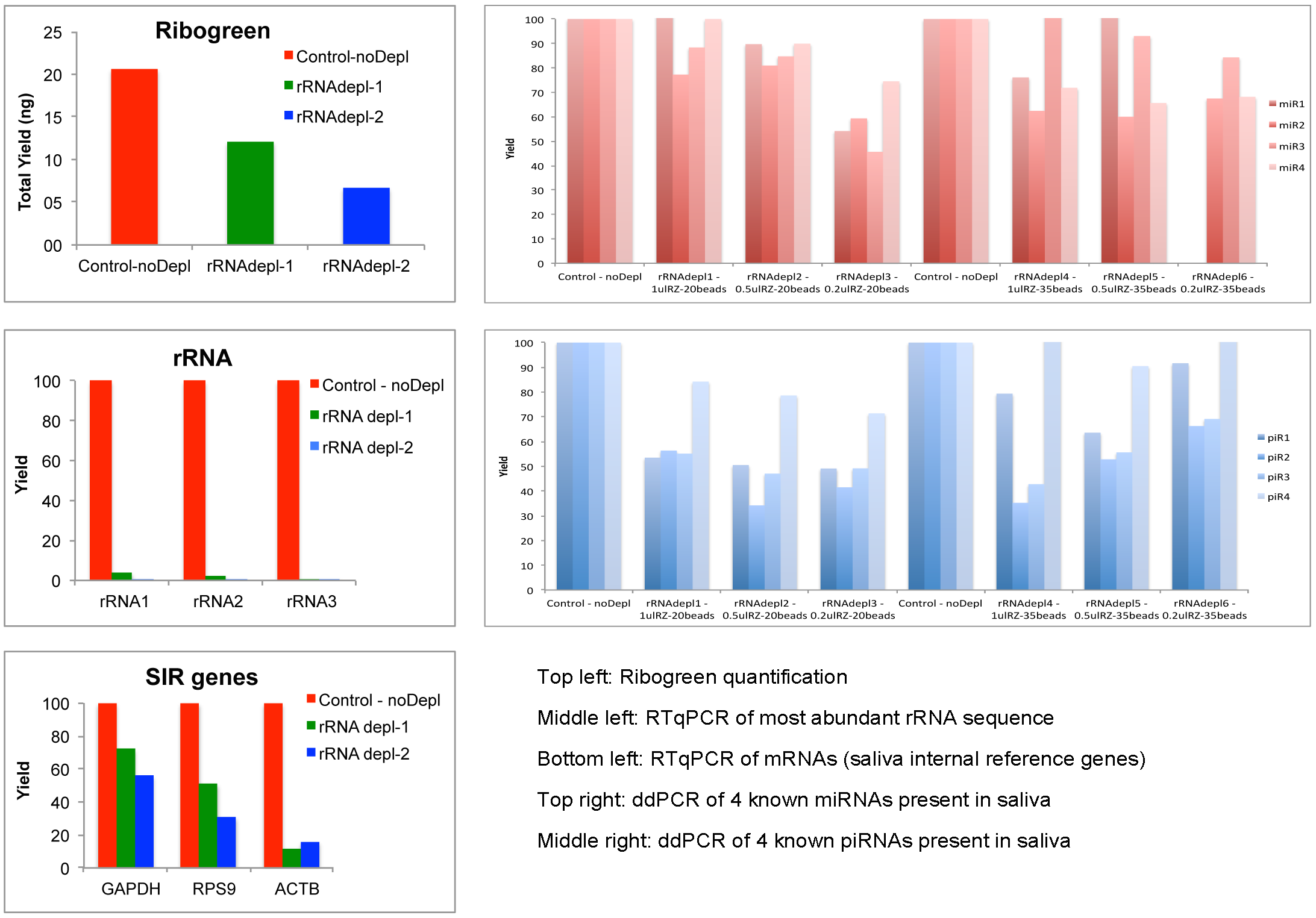
Figure 6: Total yield, qPCR/ddPCR depleted vs control samples
Share your ExRNA expertise with everyone!
MicroRNAs in cerebrospinal fluid as biomarkers for Alzheimer’s disease
There is one comment.

Jiahui Sun says:
Dear Dr. Majem,
I am very interested in what you have done in salivary exRNAs, but I am a little confused about the usage of extraction device and your modified protocol. As we all know, RNA will be rapidly degraded in the open air, so may I know how you deal with saliva extracted from the patients? Blood should be processed by centrifugation and stored in -80℃ within two hours after taking, so If possible, I want to know the most immediate operation after taking salvia from patients, and how to promise RNA in saliva stable.
Thank you so much!