Image: Kelsey Burke
This post originated as a press release from the University of Pennsylvania.
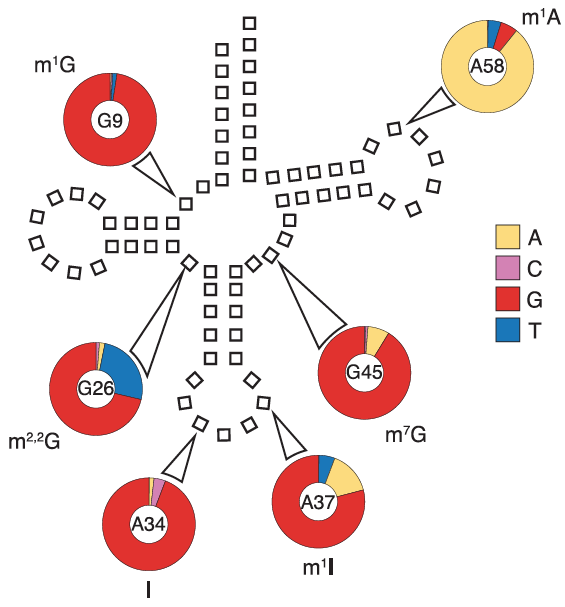
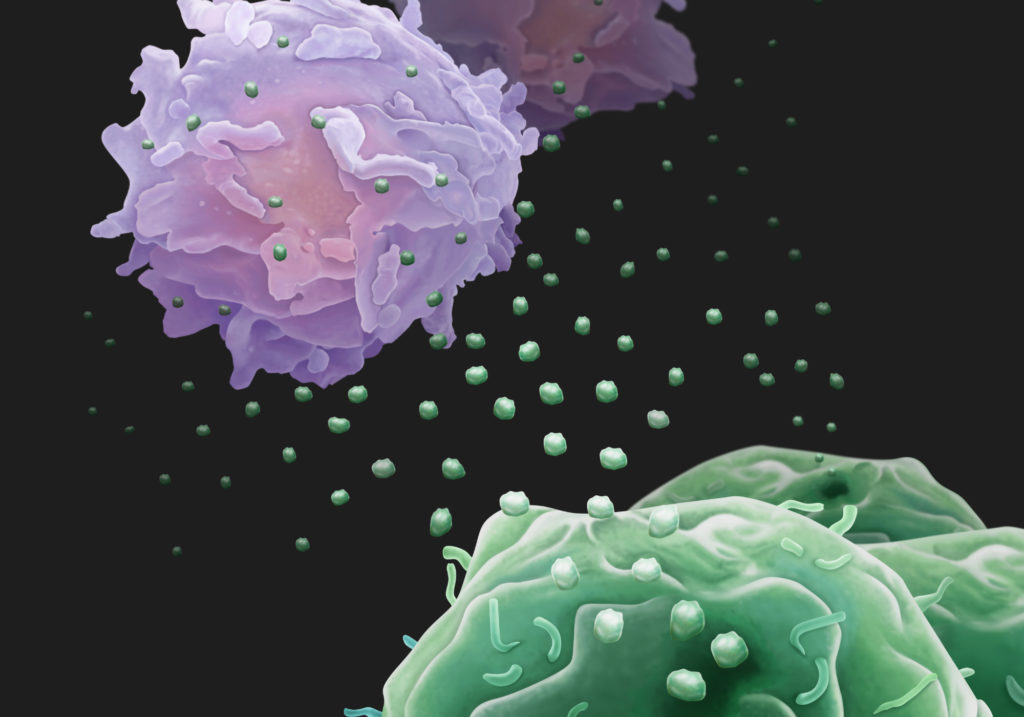
Cancerous tumors are more than a lump of cells growing out of control; they participate in active combat with the immune system for their own survival. Being able to evade the immune system is indeed a hallmark of cancer. Now, researchers from the University of Pennsylvania show that, to assist in the fight, cancer cells release biological “drones,” small vesicles called exosomes circulating in the blood and armed with the protein PD-L1, which causes T cells to tire before they have a chance to reach the tumor and do battle.
The work, published in the journal Nature (Chen et al., 2018), is a collaboration between Wei Guo of Penn’s School of Arts and Sciences and Xiaowei Xu of the Perelman School of Medicine. While primarily focused on metastatic melanoma, the team found that breast and lung cancer also release the PD-L1-carrying exosomes.
The research offers a paradigm-shifting picture of how cancers take a systemic approach to suppressing the immune system. In addition, it also points to a new way to predict which cancer patients will respond to certain checkpoint inhibitor drugs, which disrupt immune suppression to fight tumors, and a means of tracking the effectiveness of such therapies.
“Immunotherapies are life-saving for many patients with metastatic melanoma, but about 70 percent of these patients don’t respond,” says Guo, a professor of biology. “These treatments are costly and have toxic side effects, so it would be very helpful to know which patients are going to respond. Identifying a biomarker in the bloodstream could potentially help make early predictions about which patients will respond and, later on, could offer patients and their doctors a way to monitor how well their treatment is working.”
“Exosomes are tiny lipid-encapsulated vesicles with the diameter less than one-hundredth of a red blood cell. What we have found with these circulating exosomes is truly remarkable,” says Xu. “We collected blood samples from melanoma patients treated with anti-PD1 checkpoint inhibitor therapy. This type of liquid biopsy assay allows us to monitor tumor-related immune suppression with time.”
One of the most successful innovations in cancer therapy has been the use of checkpoint inhibitor drugs, which are designed to block attempts by cancer cells to suppress the immune system to allow tumors to thrive and spread. One of the primary targets for this class of drugs is PD-1, a protein on the surface of T cells. Tumor cells express PD-L1, which interacts with PD-1, effectively turning off that cell’s anti-cancer response. Blocking that interaction using checkpoint inhibitors reinvigorates T cells, allowing them to unleash their cancer-killing power on the tumor.
While it was known that cancer cells carried PD-L1 on their surface, Guo, Xu and colleagues found, in the new work, that exosomes from human melanoma cells also carried PD-L1 on their surface. Exosomal PD-L1 can directly bind to and inhibit T cell functions. Identifying the exosomal PD-L1 secreted by tumor cells provides a major update to the immune checkpoint mechanism, and offers novel insight into tumor immune evasion.
“Essentially exosomes secreted by melanoma cells are immunosuppressive.” Guo says. “We propose a model in which these exosomes act like drones to fight against T cells in circulation, even before the T cells get near to the tumor.”
Since a single tumor cell is able to secrete many copies of exosomes, the interaction between the PD-L1 exosomes and T cells provides a systemic and highly effective means to suppress anti-tumor immunity in the whole body. This may help explain why cancer patients have weakened immune systems.
Because exosomes circulate in the bloodstream, they present an accessible way of monitoring the cancer-T cell battle through a blood test, compared to a traditional, more-invasive tumor biopsy. After an acute phase of treatment, the researchers envision such a test as a way to monitor how well the drugs are keeping cancer cells in check.
By measuring pre-treatment levels of PD-L1, oncologists may be able to predict the extent of tumor burden in a patient and associate that with treatment outcome. In addition, a blood test could measure the effectiveness of a treatment. For example, levels of exosomal PD-L1 could indicate the level of T cell invigoration by immune checkpoint inhibitors.
“In the future, I think we will begin to think about cancers as a chronic disease, like diabetes,” says Guo. “And just as diabetes patients use glucometers to measure their sugar levels, it’s possible that monitoring PD-L1 and other biomarkers on the circulating exosomes could be a way for clinicians and cancer patients to keep tabs on treatment. It’s another step toward precision and personalized medicine.”
Reference
Chen G. et al. Exosomal PD-L1 contributes to immunosuppression and is associated with anti-PD-1 response. Nature (2018) 560: 382–386. doi: 10.1038/s41586-018-0392-8 PMID: 30089911
Guo and Xu coauthored the work with Penn’s Gang Chen, Alexander C. Huang, Wei Zhang, Min Wu, Jiegang Yang, Beike Wang, Honghong Sun, Wenqun Zhong, Bin Wu, Xiaoming Liu, Lei Guan, Tin Li, Shujing Liu, Ruifeng Yang, Youtao Lu, Liyun Dong, Suzanne McGettigan, Ravi Radhakrishnan, Junhyong Kim, Youhai H. Chen, Giorgos C. Karakousis, Tara C. Gangadhar, Lynn M. Schuchter, and E. John Wherry, as well as collaborators from Wuhan University, The Wistar Institute, Xi’an Jiaotong University, the University of Texas MD Anderson Cancer Center, and the Mayo Clinic.
The research was supported by the National Institutes of Health (GM111128, GM085146, AI105343, AI108545, AI082630, and AI117950), Parker Institute for Cancer Immunotherapy, American Heart Association, Tara Miller Melanoma Foundation, University of Pennsylvania, Wistar Institute, Dr. Miriam and Sheldon G. Adelson Medical Research Foundation, CAST Foundation, and NSFC Foundation.
Wei Guo is a professor of biology in the School of Arts and Sciences, and Xiaowei Xu is a professor of pathology and laboratory medicine and of dermatology in the Perelman School of Medicine at the University of Pennsylvania.
Wei Guo, Xiaowei Xu, and Gang Chen are listed as inventors on a patient owned by the University of Pennsylvania related to this work. Guo and Xu serve on the Scientific Advisory Board and have equities in Exo Bio, a company that has licensed the patent from the University of Pennsylvania.