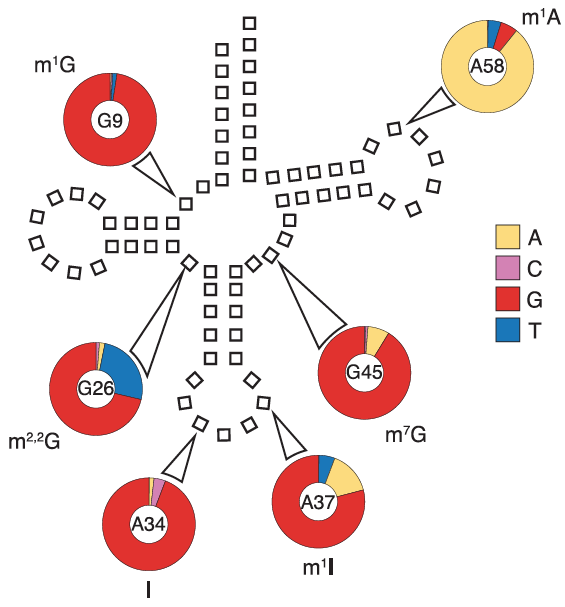
![]()
This blog post was adapted from a press release by the Baylor College of Medicine. See this related video from the ERCC Webinar Series for a discussion of the exceRpt pipeline used in the analysis presented here.
Scientists have improved their understanding of a new form of cell-cell communication that is based on extracellular RNA (exRNA). RNA, a molecule that was once thought to function only inside cells, is now known to participate in a cell-cell communication system that delivers messages throughout the body. To better understand this system, the Extracellular RNA Communication Consortium (ERCC), which includes researchers from Baylor College of Medicine, created the exRNA Atlas resource, the first detailed catalog of exRNAs in human bodily fluids. They also developed web-accessible computational tools other researchers can use to analyze exRNAs from their own data. The study (Murillo, Thistlethwaite, et al. 2019), published in the journal Cell, contributes the first ‘map of the terrain’ that will enable scientists to study the potential roles exRNA plays in health and disease.
“About 10 years ago, scientists began discovering a new communication system between cells that is mediated by exRNA,” said corresponding author Dr. Aleksandar Milosavljevic, professor of molecular and human genetics and co-director of the Computational and Integrative Biomedical Research Center at Baylor College of Medicine. “The system seems to work in normal physiological conditions, as well as in diseases such as cancer.”
The Milosavljevic lab worked with other members of the ERCC to analyze human exRNAs from 19 studies. They soon realized that the system was significantly more complex than initially assumed. Due to that unanticipated complexity, existing laboratory methods failed to reproducibly isolate exRNAs and their carriers. To help create the first map of this complex system of communication, Milosavljevic and his colleagues used computational tools to deconvolute the complex experimental data. Deconvolution refers to a mathematical method and a computational algorithm that separates complex information into components that are easier to interpret.
“Using computational deconvolution, we discovered six major types of exRNA cargo and their carriers that can be detected in bodily fluids, including serum, plasma, cerebrospinal fluid, saliva, and urine,” said co-first author Oscar D. Murillo, a graduate student in Baylor’s Molecular and Human Genetics Graduate Program working in the Milosavljevic lab. “The carriers act like molecular vessels moving their RNA cargo throughout the body. They include lipoproteins – one of the major carriers is High-Density Lipoprotein (HDL or the “good cholesterol”) – a variety of small protein-containing particles, and small vesicles, all of which can be taken up by cells.”
The researchers found that the computational method helps reveal biological signals that could not previously be detected in individual studies due to the naturally complex variation in the biological system. For example, in an exercise challenge study their computational approach revealed differences before and after exercise in the proportions of the exRNA cargo in HDL particles and vesicles in human plasma.
“Exercise increased a proportion of RNA molecules involved in regulating metabolism and muscle function, suggesting adaptive response of the organism to exercise challenge,” Milosavljevic said. “This finding opens the possibility that in other conditions, both in health or disease, the computational method might identify signals that could have physiological and clinical relevance.”
To help researchers around the world with their analyses, Murillo, Milosavljevic and their colleagues have made a computational tool available online (https://exRNA-Atlas.org).
“We anticipate that it will take a combination of scientific knowledge, enhanced experimental techniques to isolate cargo and carriers in bodily fluids, and advanced computational methods to deconvolute and interpret the complexity of the exRNA communication system,” Murillo said.
Other contributors to this work from Baylor College of Medicine include William Thistlethwaite, Matthew E. Roth, Sal Lakshmi Subramanian, Rocco Lucero, Neethu Sha, and Andrew R. Jackson. See the full article for details about the numerous other contributors from the consortium.
This work is part of the NIH Extracellular RNA Communication Consortium paper package and was supported by the NIH Common Fund Extracellular RNA Communication Program (grant U54 DA036134).
Reference
Murillo OD, Thistlethwaite W, et al. exRNA Atlas analysis reveals distinct extracellular RNA cargo types and their carriers present across human biofluids. (2019) Cell 177:463-477. doi: 10.1016/j.cell.2019.02.018. PMID: 30951672.