| This blog is a repost of the NIH Directors’ blog from April 4th, 2023. |
|
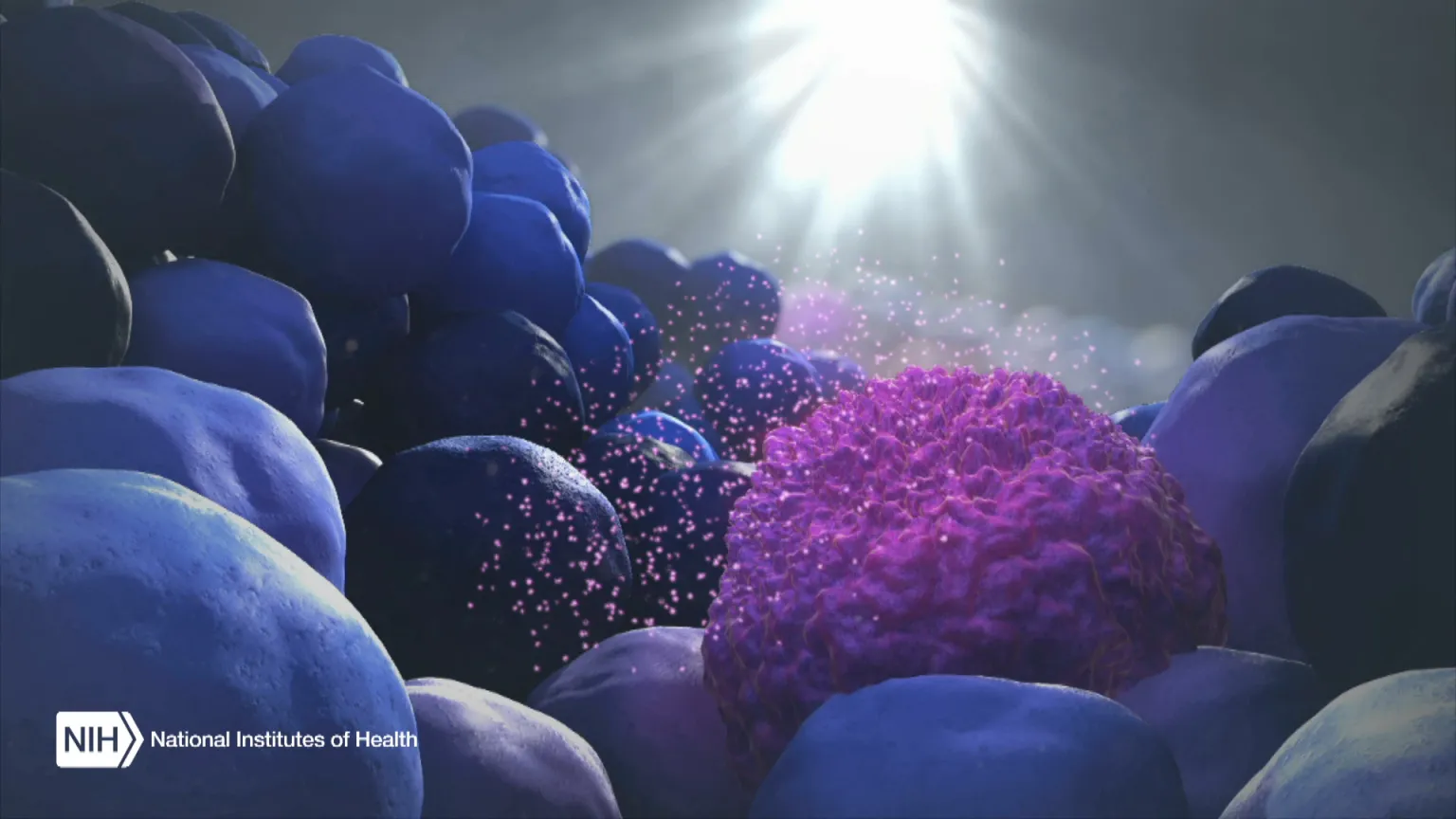
Figure Credit: XVIVO Scientific Animation, Wethersfield, CT
With just a blood sample from a patient, a promising technology has the potential to accurately diagnose non-small cell lung cancer (NSCLC), the most-common form of the disease, more than 90 percent of the time. The same technology can even predict from the same blood sample whether a patient will respond well to a targeted immunotherapy treatment.
This work is a good example of research supported by the NIH Common Fund. Many Common Fund programs support development of new tools that catalyze research across the full spectrum of biomedical science without focusing on a single disease or organ system.
The emerging NSCLC prediction technology was developed as part of our Extracellular RNA Communication Program. The program develops technologies to understand RNA circulating in the body, known as extracellular RNA (exRNA). These molecules can be easily accessed in bodily fluids such as blood, urine, and saliva, and they have enormous potential as biomarkers to better understand cancer and other diseases.
When the body’s immune system detects a developing tumor, it activates various immune cells that work together to kill the suspicious cells. But many tumors have found a way to evade the immune system by producing a protein called PD-L1.
Displayed on the surface of a cancer cell, PD-L1 can bind to a protein found on immune cells with the similar designation of PD-1. The binding of the two proteins keeps immune cells from killing tumor cells. One type of immunotherapy interferes with this binding process and can restore the natural ability of the immune system to kill the tumor cells.
However, tumors differ from person to person, and this form of cancer immunotherapy doesn’t work for everyone. People with higher levels of PD-L1 in their tumors generally have better response rates to immunotherapy, and that’s why oncologists test for the protein before attempting the treatment.
Because cancer cells within a tumor can vary greatly, a single biopsy taken at a single site in the tumor may miss cells with PD-L1. In fact, current prediction technologies using tissue biopsies correctly predict just 20 – 40 percent of NSCLC patients who will respond well to immunotherapy. This means some people receive immunotherapy who shouldn’t, while others don’t get it who might benefit.
To improve these predictions, a research team led by Eduardo Reátegui, The Ohio State University, Columbus, engineered a new technology to measure exRNA and proteins found within and on the surface of extracellular vesicles (EVs) [1]. EVs are tiny molecular containers released by cells. They carry RNA and proteins (including PD-L1) throughout the body and are known to play a role in communication between cells.
As the illustration above shows, EVs can be shed from tumors and then circulate in the bloodstream. That means their characteristics and internal cargo, including exRNA, can provide insight into the features of a tumor. But collecting EVs, breaking them open, and pooling their contents for assessment means that molecules occurring in small quantities (like PD-L1) can get lost in the mix. It also exposes delicate exRNA molecules to potential breakdown outside the protective EV.
The new technology solves these problems. It sorts and isolates individual EVs and measures both PD-1 and PD-L1 proteins, as well as exRNA that contains their genetic codes. This provides a more comprehensive picture of PD-L1 production within the tumor compared to a single biopsy sample. But also, measuring surface proteins and the contents of individual EVs makes this technique exquisitely sensitive.
By measuring proteins and the exRNA cargo from individual EVs, Reátegui and team found that the technology correctly predicted whether a patient had NSCLC 93.2 percent of the time. It also predicted immunotherapy response with an accuracy of 72.2 percent, far exceeding the current gold standard method.
The researchers are working on scaling up the technology, which would increase precision and allow for more simultaneous measurements. They are also working with the James Comprehensive Cancer Center at The Ohio State University to expand their testing. That includes validating the technology using banked clinical samples of blood and other bodily fluids from large groups of cancer patients. With continued development, this new technology could improve NSCLC treatment while, critically, lowering its cost.
The real power of the technology, though, lies in its flexibility. Its components can be swapped out to recognize any number of marker molecules for other diseases and conditions. That includes other cancers, neurodegenerative diseases, traumatic brain injury, viral diseases, and cardiovascular diseases. This broad applicability is an example of how Common Fund investments catalyze advances across the research spectrum that will help many people now and in the future.
Reference
[1] Nyugen LTH et al. An immunogold single extracellular vesicular RNA and protein (AuSERP) biochip to predict responses to immunotherapy in non-small cell lung cancer patients J Extracell Vesicles (2023) 11: e12258. doi: 10.1002/jev2.12258 PMID: 36093740.
Links
Extracellular RNA Communication Program (ERCC) (Common Fund)
Video: Unlocking the Mysteries of Extracellular RNA Communication
Upcoming Meeting: ERCC19 Research Meeting (May 1-2, 2023)
Eduardo Reátegui Group for Bioengineering Research (The Ohio State University College of Engineering, Columbus)
Note
Dr. Lawrence Tabak, who performs the duties of the NIH Director, has asked the heads of NIH’s Institutes, Centers, and Offices to contribute occasional guest posts to the blog to highlight some of the interesting science that they support and conduct. This is the 27th in the series of NIH guest posts that will run until a new permanent NIH director is in place.



