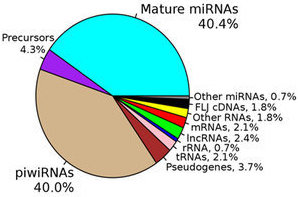
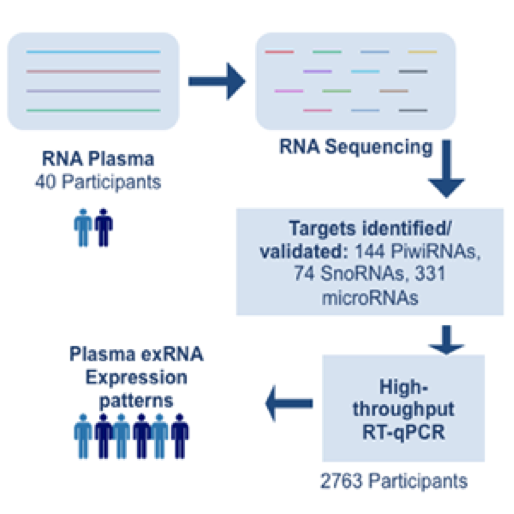
The ability to perform small RNA sequencing on extracellular RNA samples allows us to measure, at least in a semi-quantitative manner, the abundance of miRNAs, piRNAs, fragments of tRNAs, long non-coding RNAs, and coding RNAs. One of the most significant advantages of RNA-Seq technology is that it can detect and measure any RNA that is present, whether or not it is a known sequence. Nonetheless, there are important unanswered questions about the accuracy of RNA-Seq and the optimal approach for processing the data obtained. All RNA-seq experiments are subject to sources of systematic variation such as library size, transcript length, and G-C content (Dillies et al., 2013). Small RNA-seq experiments are further impacted by the highly non-normal distribution of expression of different small RNAs, particularly miRNAs. Often a few miRNAs account for a very large fraction of total reads while the vast majority of miRNAs each contribute a small percentage of reads. Moreover, sample input amounts of extracellular RNA are often extremely limited, increasing the potential for both sampling error and experimental bias.
To address these issues, a number of normalization methods have been developed which can be assigned three basic categories: 1) scaling; 2) normalizing in order to achieve similar data distributions; and 3) Reads Per Kilobase Mapped (RPKM). There appears to be a lack of consensus regarding the optimal normalization method, but it is known that different methods can result in different results in downstream analysis, particularly differential expression analysis (DE).
Two studies highlighted here, Garmire and Subramaniam, 2012 and Dillies et al., 2013, do not agree on the best way to do normalization for small RNA-seq, but point to a number of methods analyzed and elucidate some of the key problems. We review these studies briefly here and point out the importance of rigorous comparisons of normalization methods for the future of differential comparisons of data sets.
The first class of methods, scaling methods, involves application of a standard linear mathematical operation to each sample. Scaling generally means changing the size of something, and normalizing by simply dividing by the total read number (going from read numbers to read fractions) is probably the simplest kind of scaling. Scaling approaches to normalization include global scaling, Lowess, and the Trimmed Mean Method (TMM) (Robinson and Oshlack, 2010). These approaches each use a different method to calculate a linear scaling factor. Global scaling uses a factor that is based on the difference in the means of the data sets to be compared. Lowess normalizes based on a multiple-regression model. TMM determines a scaling factor, which is the weighted trimmed mean of log expression. This factor is calculated after double trimming values at the two extremes based on log-intensity ratios (M-values) and log-intensity averages (A-values). According to Garmire and Subramaniam, variability in estimated RNA content may be even more pronounced for microRNA-seq datasets after application of this method.
Another general approach to normalization is to preserve aspects of the distribution of the data among different data sets. As with scaling, there are a number of different approaches to achieving matched data distributions, including quantile, Variance stabilization (VSN), the invariant method (INV), and DESeq. Quantile normalization has been extensively used for microarray data, with the goal of making the distribution of expression levels across samples similar. Conditional quantile normalization is a modification of quantile normalization that combines robust generalized regression and quantile normalization (Hansen, Irizarry, and Wu, 2012). The goal of the VSN method is to make the distribution of variance across different levels of expression similar. In INV normalization, a set of invariant miRNAs are selected, which are then used with one of the other methods (such as Lowess or VSN). In DESeq, a scaling factor for each sample in the dataset is obtained by computing the median of the ratios of each gene in one sample over the geometric mean of that gene across all samples. The same scaling factor is then applied to the read counts for all of the genes in that sample. RPKM is a method that has been widely used for long RNAseq datasets. RPKM is performed on each sample separately, and consists of taking the ratio of the number of counts for a given gene and the product of the total counts for all genes and the mature transcript length for that gene. Methods such as DEseq and TMM rely on the assumption that most genes are not differentially expressed (Dillies et al., 2013), which may not hold true for all microRNA-seq data sets.
Garmire and Subramaniam compared a number of normalization methods applied to mammalian microRNA-Seq data using two publicly available datasets that were chosen by the authors due to the availability of matched PCR data. They assessed the performance of the normalization methods by calculating the mean square error (MSE) and the Kolmogorv-Smirnov statistic (which is a measure of the difference of two distributions), as well as comparison with PCR data on the same samples and inspection of the results of differential expression. The authors show that Lowess, quantile and VSN normalization resulted in a smaller MSE, while TMM and VSN produced a higher MSE. Similarly, TMM and INV resulted in a larger K-S statistic, indicating a bigger change in the distribution. Quantile and Lowess normalization also had the best concordance with qPCR data. The authors thus concluded that Lowess and quantile normalization performed better than other methods studied. The primary limitations of this study include the fact that it did not incorporate strategies to compare the results of the normalization method to any “gold standard” for which the real distribution was known, and that the number of data sets used for the analysis was very small. In addition, other methods have been implemented since the publication of their study.
Dillies et al. published another comparison of normalization methods for RNAseq data in about the same time frame. Most of the datasets used were long RNAseq data, but a murine microRNA dataset was included. The seven methods compared in this study included TC (total counts), UQ (Upper Quartile), Median, DEseq, TMM, quantile and RPKM. TC, UQ and median are scaling approaches that are quite similar, involving the calculation of a scaling factor based on either the ratio of total counts (TC), upper quartile of counts (UQ) or median of counts. The authors assessed these normalization methods on real as well as simulated data. In their analysis, it is apparent that when boxplots of counts before and after normalization are assessed, the differences are most apparent in conditions where there are large differences in library size between samples. These differences do not improve after TC and RPKM normalization of the microRNA-seq data. Other features of microRNA-seq data that influence the results of the normalization according to the authors are the presence of high-count genes and a large number of 0 counts. Quantile normalization increases the intra-condition variability in the murine miRNA data. Based on the results of differential expression in this study, it is apparent that the differences in the final results depend on the normalization method and not on the model chosen to assess for differential expression (DESeq or TSPM). In the simulated data, which contains high count genes and may resemble microRNA seq data more closely than the other datasets, only DESeq and TMM were successful in achieving a low false positive rate and high power. The authors conclude that DESeq and TMM are the methods of choice based on their ability to perform in the presence of different library sizes and composition.
The differences in the conclusions from these two studies are likely due to the different normalization methods and specific datasets each used, and on the parameters they used to evaluate the performance of the normalization methods. Since the Garmire and Subramanian paper looks only at miRNA data and the Dillies et al. paper looks at a mix of data, including miRNA and mRNA data, it is difficult to compare the results. We suggest that the additional complexities of compiling mRNA levels from multiple read sequences may confuse the latter analysis. However, both studies agree that the use of different normalization approaches can result in significant differences in downstream differential expression results. One of the key weaknesses of both of these papers is the lack of data from “gold standard” datasets for which the quantities of the different RNAs were definitively known.
For this reason, it would be valuable to develop the tools necessary for rigorous comparison of the available normalization methods. Such tools may include the generation of standardized data sets, such as small RNA libraries constructed from purely synthetic miRNAs, for which the content can be completely controlled, or RNAs from biological specimens that contain synthetic miRNAs spiked in at controlled concentrations. Corresponding qPCR results, used with calibration curves, should be generated for such datasets, which will serve as an orthogonal measurement technology for developing and evaluating normalization methods. Overall, this important topic needs careful attention for the establishment of reference exRNA profiles, and for the realization of the full potential of the powerful technology of high throughput RNA-seq.
* * *
Note that one of us recently presented a web seminar on a related topic, Understanding and using small RNA-seq, that is available for viewing. Also, members of the ERCC will be jointly presenting a workshop on Data Normalization Challenges and Solutions as part of the CHI conference Extracellular RNA in Drug and Diagnostic Development in Cambridge, MA, 3-6 April 2016. See the Event page for more details.