Therapeutic exosomes and Huntington’s disease
Extracellular vesicles, specifically exosomes, are currently being explored as therapeutic delivery systems for disease-targeting RNA molecules. In a talk at the ERCC9 conference, Reka A. Haraszti, M.D., a researcher in Dr. Anastasia Khvorova’s group at the University of Massachusetts Medical School, described how exosomes could be used to treat Huntington’s disease, a progressive neurodegenerative disorder. There are currently no effective therapies for this illness, which is caused by a mutation in the Huntingtin gene. Exosomes capable of transporting molecular payloads designed to silence the defective Huntingtin gene represent a potential therapy for this fatal disease.
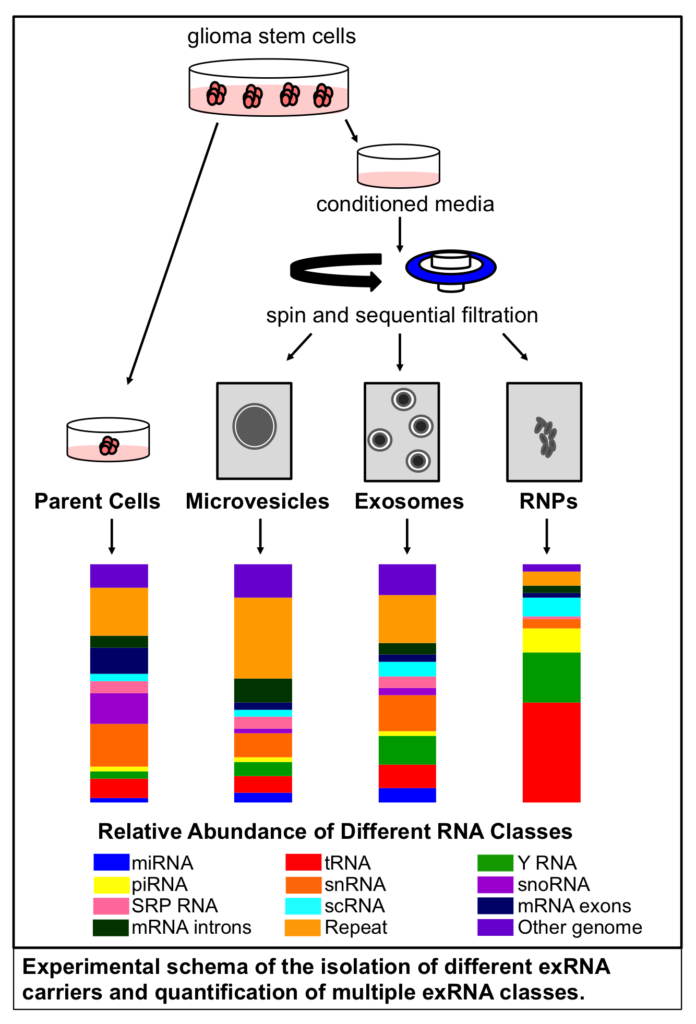
Comparison of exosome production methods
Technical challenges in the large-scale production of exosomes currently limit their utility for disease treatment. To address this issue, Dr. Haraszti and Dr. Khvorova’s group teamed up with MassBiologics to develop and compare two different exosome production methods for yield and therapeutic efficacy of the exosomes. They utilized Tangential Flow Filtration (TFF) and ultracentrifugation to isolate exosomes from the conditioned media of cultured mesenchymal stem cells.
In TFF, conditioned media is continuously swept along the surface of a filter while a downward pressure is applied to force molecules through the filter. This process is like shaking a sifter to concentrate large particles blocking the holes in the filter, allowing smaller particles to pass through. In contrast, ultracentrifugation works by placing the conditioned media in a column of viscous fluid and spinning rapidly to separate extracellular vesicles in the media by their differing densities.
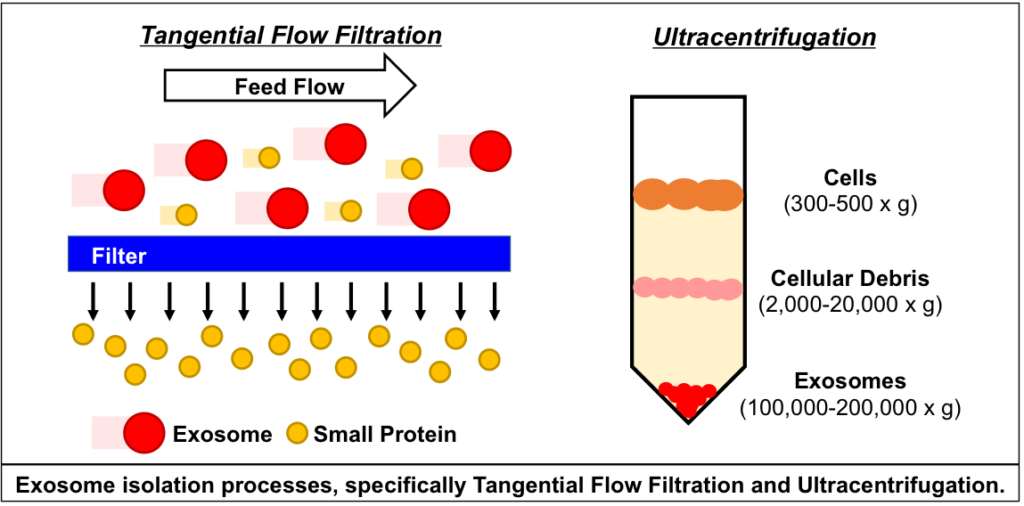
The researchers found that isolation by TFF resulted in 10-100 times more exosomes than ultracentrifugation. TFF-generated exosomes were also more heterogenous and contained 10 times more protein.
Exosomes produced by TFF and ultracentrifugation were also studied for their therapeutic potential in Huntington’s disease. After purification, exosomes were loaded with small interfering RNA (siRNA) molecules that could silence the expression of the mutant Huntingtin gene in target cells that take up the exosomes.
In cell cultures of primary neurons, TFF-generated exosomes showed greater inhibition of Huntingtin expression than those generated by ultracentrifugation. Moreover, TFF-generated exosomes infused into the brains of mice suppressed Huntingtin gene expression in vivo.
Future for therapies using exosomes isolated by Tangential Flow Filtration
The findings of Dr. Haraszti’s group indicate that Tangential Flow Filtration can generate a higher yield of exosomes for clinical use than older methods. Further, TFF-generated exosomes were effective gene therapy agents in experimental models of Huntington’s disease, and hold promise as delivery systems for clinical treatments.
Related Mini-conference
There is an upcoming mini-conference on EV manufacturing and isolation, a topic closely related to the research described here. The in-person conference is in Gainesville, Florida, but a webcast will also be available for those who want to participate remotely.
References
1. Haraszti RA, et al. Loading of extracellular vesicles with chemically stabilized hydrophobic siRNAs for the treatment of disease in the central nervous system. Bio-protocol (2017) 7: e2338. doi: 10.21769/BioProtoc.2338
2. Schwartz L., and Seeley K. Introduction to tangential flow filtration for laboratory and process development applications. Retrieved from https://laboratory.pall.com/content/dam/pall/laboratory/literature-library/non-gated/id-34212.pdf
3. Sunkara V, Woo HK, and Cho YK. Emerging techniques in the isolation and characterization of extracellular vesicles and their roles in cancer diagnostics and prognostics. Analyst (2016) 141: 371-81. doi: 10.1039/c5an01775k
4. Synder Filtration. “Characterization of polymeric, porous membranes: UF/MF & solute rejection measurements.” Retrieved from https://synderfiltration.com/learning-center/articles/membranes/characterization-of-polymeric-porous-membranes/