Recent research from members of the ExRNA Communication Consortium (ERCC) suggests that extracellular RNAs (exRNAs) circulating in plasma play an active role in insulin resistance (IR). Insulin resistance is an incurable but manageable syndrome where the body stops reacting efficiently to the insulin hormone, which stimulates the uptake of glucose in the blood into cells and inhibits the body from using fat for energy, resulting in high blood sugar levels. The study, by Shav et al., points to certain exRNAs, particularly miR-122 and miR-192, as indicators and active players in IR regardless of the age, sex, or BMI of a person, which suggests that they may serve as more than metabolic markers and that they perhaps have functional, trans-organ roles in mediating IR.
Previous research has demonstrated that exRNAs have different functions in pathways relating to metabolic syndrome, which is a series of conditions that increase the risk of heart disease, stroke, and diabetes. For example, there is measurable miRNA dysregulation in obesity and in the progression of cardiometabolic disease. Other miRNAs are involved in brown/white fat specification, adipose tissue inflammation (Karbiener and Scheideler, 2014), and hepatic steatosis (fatty liver disease, Becker et al., 2015). Although these studies have identified specific exRNAs and miRNA networks that also have roles in IR, they have had either small sample sizes and lack validation in large populations or have been carried out in non-human models. In order to validate these data, the authors carried out a large-scale human translational study in which they analyzed detailed obesity-related phenotypic data from over 2,500 participants (most of whom were non-diabetic) from the Framingham Heart Study (FHS), an unrelated cardiovascular disease study (Feinleib et al., 1975).
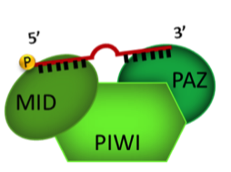
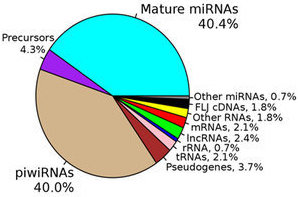
To begin, the investigators analyzed blood samples from 2,317 non-diabetic study participants and quantified the plasma extracellular circulating exRNAs. They looked at RNAs [including piwi-interacting RNA (piRNA) and small nucleolar RNA (snoRNA)] expressed above a threshold level and excluded RNAs that were not found in at least 100 participants. From the resulting panel of 391 exRNAs, the investigators identified 16 microRNAs (miRNA), 1 piRNA, and 1 snoRNA that were associated with insulin after controlling for age, sex, and BMI. Of note, the abundance of miR-122 was shown to increase in a stepwise fashion as levels of insulin increased across the population. Higher levels of both miR-122 and miR-192 in the plasma were also consistently associated with a series of metabolic phenotypes, such as greater BMI and waist circumference, visceral fat quantity and quality, and liver attenuation. On the other hand, neither miRNA was associated with subcutaneous fat. These results were consistent whether the analysis included only the non-diabetic participants or the entire FHS population.
miRNAs function to regulate gene expression, so the authors conducted a pathway analysis to determine the targets of the 16 identified miRNAs. Almost unsurprisingly, all 16 miRNAs target insulin signaling pathways such that there is ample crosstalk and targeting of multiple IR-related genes by multiple miRNAs. This analysis validated findings from previous studies that implicated several miRNA target genes in the pathogenesis of IR, notably protein tyrosine phosphatase, nonreceptor type 1 (PTP1B) (Stull et al., 2012), mitogen-activated protein kinases (MAPKs) (Wang, Goalstone & Draznin, 2004), and 5′ adenosine monophosphate-activated protein kinase (AMPK) (Ruderman et al., 2013).
For the second part of the study, the investigators determined whether the miR-122 and miR-192 associations to age, sex, and BMI held true in a separate study population, a cohort of 90 overweight or obese young adults involved in the POOL study. Analyses of the youths’ plasma samples indicated that miR-122 (but not miR-192) was associated with greater IR after adjusting for age, sex, and BMI, and that this association remained even after the miRNA was analyzed independently of age, sex, BMI, or metabolite profile.
This study provides additional evidence and translational support for the role of exRNAs in IR. The findings indicate not only an association of exRNAs with insulin levels, but that the exRNAs may be playing an active role in the development or sustainment of IR. It is therefore critical to conduct further mechanistic investigations into the role of exRNAs in the metabolic architecture of IR.